Hallazgos de la gammagrafia ósea con 99mTc-HDP en un paciente pediátrico con síndrome falciforme (Hb S/ Beta talasemia)
Estefanía López Rodríguez1, Francisco Javier García Gómez1, Rosa María Álvarez-Pérez1, Isabel Borrego Dorado1.1 - Servicio de Medicina Nuclear, Hospital Universitario Virgen del Rocío. Sevilla, España..
Descripción del caso
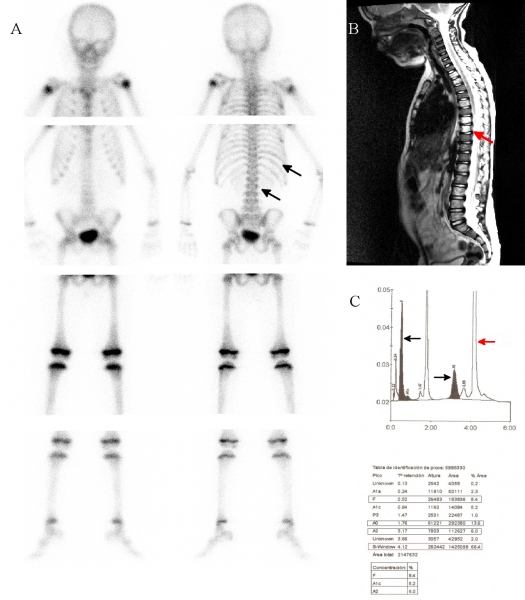
Varón de raza negra de 3 años de edad que presenta fiebre de hasta 38ºC, rigidez en el cuello, dolor abdominal y de columna cervical, así como dolores articulares de un año de evolución, que fue remitido a nuestra servicio para descartar un proceso inflamatorio / infeccioso cervical. Se realizó una gammagrafia ósea con 99mTc- hidroximetilen-difosfonato (HDP) (Figura 1A) que reveló captación irregular del radiotrazador en arcos costales y columna dorsal, hallazgos compatibles con afectación ósea isquémica. En la resonancia magnética (RM) se apreció hiperintensidad de la médula ósea, siguiendo el patrón observado en la gammagrafia (Figura 1B). Ante la sospecha de un sindrome falciforme, se realizó un estudio de hemoglobinas mediante electroforesis (Figura 1C) que confirmó la presunción diagnóstica (HbS/beta Tal). HBS fue de 66,4%, mientras que se observó niveles bajos de HbA2 y HbF (6% y 8,4% respectivamente) y un mayor nivel de HbA0 (13,6%). El estudio genético de los padres fue a su vez positivo.
Comentarios
La HbS-beta talasemia predomina en los países en los que ha habido inmigración de poblaciones con alta prevalencia, alcanzando una incidencia de 0,395 / 1000 nacimientos en la región europea(1). En pacientes con Hb S-beta talasemia, los infartos óseos múltiples son frecuentes durante el curso de la enfermedad, de modo que la gammagrafía ósea con 99mTc-HDP es útil para el clínico en el diagnóstico diferencial entre infartos óseos asépticos y osteomielitis secundaria(2). Una gammagrafía complementaria con coloide podría ayudar a demostrar infartos del bazo, que generalmente ocurren después de la primera década de la vida(3).
Referencias
- Modell B, Darlison M, Birgens H, et al. Epidemiology of haemoglobin disorders in Europe: an overview. Scand J Clin Lab Invest. 2007;67:39-69.
- Mikosch P, Jauk B, Kaulfersch W, et al. Scintigraphic findings in a patient with sickle-cell thalassemia and reccurent pain attacks. Wien Med Wochenschr 2003; 3-4:83-8.
- Ahmed H. Al-Salem. Splenic complications of sickle cell anemia and the role of splenectomy. ISRN Hematol. 2011; 2011: 864257.